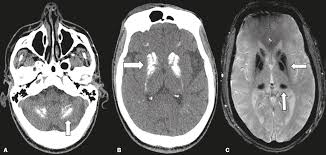
Calcificações intracranianas neonatais e diagnóstico diferencial com infecções congênitas (pseudo-TORCH) Sobre o artigo Trata-se de um relato clínico publicado na seção Images in Neonatal Medicine, descrevendo um recém-nascido masculino, termo, nascido por cesariana de emergência devido a sofrimento fetal e líquido meconial, admitido em unidade neonatal de cuidados intermediários para oxigenoterapia de alto fluxo. Era filho único de pais consanguíneos de segundo grau, com pré-natal sem intercorrências. Os exames iniciais evidenciaram trombocitopenia importante, linfocitose leve e alterações de função hepática. A ultrassonografia craniana demonstrou calcificações cerebrais difusas. Inicialmente, foi estabelecida hipótese diagnóstica de infecção congênita do grupo TORCH (toxoplasmose, rubéola, citomegalovírus, herpes simples), sendo realizada investigação específica. Contudo, a triagem estendida para TORCH foi negativa. Diante da persistência do quadro neurológico — hipotonia central e fraqueza significativa do reflexo de sucção — e após discussão com centro de referência em neurologia, foi considerada a hipótese de síndrome de Aicardi-Goutières (AGS), posteriormente confirmada por sequenciamento genômico. Métodos utilizados Avaliação clínica neonatal completa. Exames laboratoriais iniciais: hemograma (trombocitopenia, linfocitose) e testes de função hepática. Ultrassonografia craniana, que evidenciou calcificações parenquimatosas e periventriculares difusas, com possível agenesia do corpo caloso. Triagem estendida para infecções congênitas do grupo TORCH. Sequenciamento rápido do genoma completo em trio (recém-nascido e ambos os pais). O sequenciamento identificou mutação patogênica homozigótica no gene TREX1, herdada biparentalmente, confirmando o diagnóstico de AGS. Resultados Os principais achados foram: Calcificações intracranianas bilaterais extensas ao ultrassom craniano, localizadas tipicamente em: Gânglios da base Substância branca Regiões periventriculares Possível agenesia do corpo caloso. Exames laboratoriais com trombocitopenia e disfunção hepática. Triagem TORCH negativa. Confirmação genética de mutação homozigótica patogênica em TREX1. Esses achados consolidaram o diagnóstico de síndrome de Aicardi-Goutières. Discussão A síndrome de Aicardi-Goutières deve ser considerada no diagnóstico diferencial das infecções congênitas do grupo TORCH. Na fase neonatal, a AGS pode apresentar fenótipo clínico e alterações laboratoriais sobreponíveis às infecções congênitas, incluindo: Alterações bioquímicas séricas Manifestações neurológicas precoces Calcificações intracranianas O artigo enfatiza que: A ultrassonografia craniana é exame fundamental na investigação de defeitos neurológicos neonatais. O padrão de calcificações pode sugerir AGS ou uma condição pseudo-TORCH. A confirmação diagnóstica depende de testes genéticos. Portanto, a diferenciação entre infecção congênita e interferonopatia monogênica como a AGS é crucial para adequada orientação prognóstica e aconselhamento genético. Conclusão A síndrome de Aicardi-Goutières é uma causa rara, porém relevante, de calcificações intracranianas no período neonatal. Deve ser incluída como diagnóstico diferencial das infecções congênitas TORCH, especialmente quando a investigação infecciosa é negativa. O ultrassom craniano tem papel central na suspeita diagnóstica, mas a confirmação definitiva é genética, particularmente por meio da identificação de mutações patogênicas como no gene TREX1. Insights clínicos Quando suspeitar de síndrome de Aicardi-Goutières no período neonatal? Deve-se suspeitar diante de calcificações intracranianas difusas associadas a alterações laboratoriais e fenótipo semelhante a infecção congênita, especialmente quando a triagem TORCH é negativa. Qual exame de imagem é fundamental na investigação inicial? A ultrassonografia craniana é essencial e pode demonstrar calcificações bilaterais em gânglios da base, substância branca e regiões periventriculares. Quais alterações laboratoriais podem estar presentes? Trombocitopenia, linfocitose leve e alterações da função hepática, mimetizando infecção congênita. Como se confirma o diagnóstico? Por meio de teste genético, como o sequenciamento do genoma completo, com identificação de mutação patogênica (neste caso, mutação homozigótica em TREX1). Por que é importante diferenciar AGS de infecção TORCH? Porque o manejo, o prognóstico e o aconselhamento genético diferem significativamente, sendo a AGS uma interferonopatia monogênica e não uma infecção congênita. Para ver mais conteúdos como este, acesse: NeoPed Hub
Faça login para acessar o conteúdo
ou cadastre-se. | ESQUECI MINHA SENHA