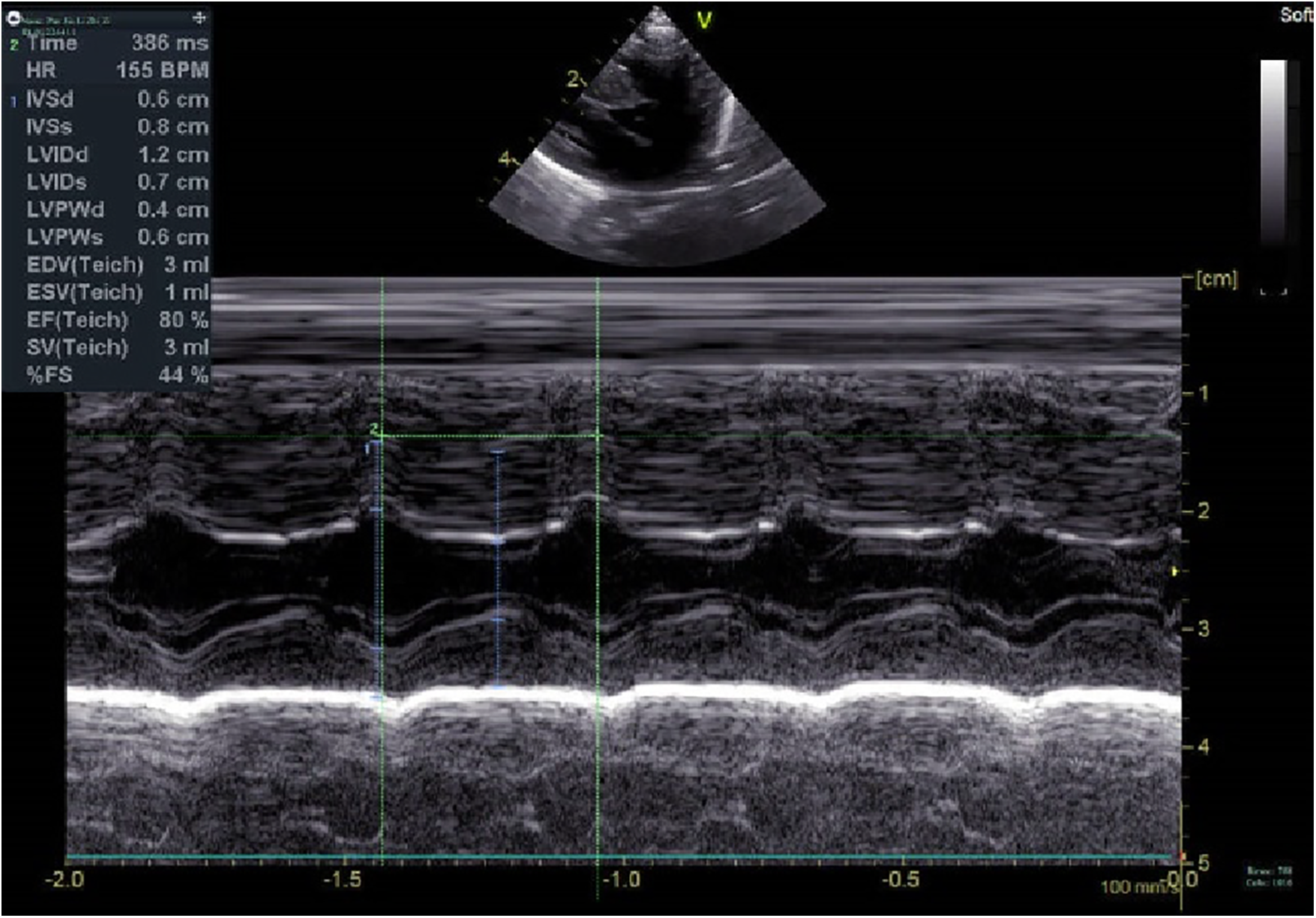
Cardiomiopatia hipertrófica neonatal com dispneia como primeiro sintoma: relato de caso Sobre o artigo O artigo descreve um caso raro de cardiomiopatia hipertrófica (CMH) diagnosticada no período neonatal, destacando sua apresentação inicial com dispneia. A CMH é caracterizada por hipertrofia miocárdica, disfunção diastólica e risco elevado de arritmias e morte súbita. A doença apresenta grande heterogeneidade clínica e genética, sendo de difícil diagnóstico precoce, especialmente em neonatos. A história familiar e a investigação genética têm papel fundamental na identificação precoce e no prognóstico. Métodos utilizados Trata-se de um relato de caso clínico com acompanhamento longitudinal desde o período neonatal até os 4 anos de idade. Foram utilizados: Avaliação clínica seriada Ecocardiografia periódica Eletrocardiograma Radiografia de tórax Testes laboratoriais Sequenciamento genético familiar (incluindo pais e parentes próximos) Monitoramento terapêutico com betabloqueador (metoprolol) Intervenção por ablação septal percutânea guiada por imagem O estudo seguiu as diretrizes CARE para relatos de caso. Resultados Recém-nascido prematuro (31 semanas) apresentou dispneia precoce e foi diagnosticado com CMH não obstrutiva por ecocardiografia. Evolução progressiva com hipertrofia do septo interventricular (de 6 mm ao nascimento para até 15,4 mm durante seguimento, conforme tabela na página 3). Desenvolvimento de sinais clínicos ao longo do tempo: dificuldade alimentar, taquipneia, sudorese e baixo ganho ponderal. Progressão para forma obstrutiva grave com risco elevado de insuficiência cardíaca, arritmias e morte súbita. Tratamento inicial com metoprolol não impediu progressão da doença. Aos 4 anos, paciente foi submetido à ablação septal percutânea com melhora ecocardiográfica e clínica. Teste genético identificou mutação patogênica no gene MYBPC3 (c.821+1G>A), presente também em familiares, confirmando etiologia hereditária. Discussão A CMH pediátrica é rara, mas associada a alta morbimortalidade, especialmente quando de início precoce. A apresentação neonatal é frequentemente inespecífica, podendo estar associada a prematuridade e desconforto respiratório, o que dificulta o diagnóstico. A ecocardiografia é o principal método diagnóstico, permitindo identificar hipertrofia, obstrução do trato de saída do ventrículo esquerdo e sinais como movimento anterior sistólico da valva mitral (SAM). Aspectos genéticos são centrais: Cerca de 60% dos casos têm origem hereditária Genes sarcoméricos, especialmente MYH7 e MYBPC3, são os mais frequentemente envolvidos A expressão fenotípica é variável, influenciada por fatores genéticos e ambientais Apesar do tratamento clínico com betabloqueadores, o caso demonstrou progressão da doença, exigindo intervenção invasiva. A ablação septal mostrou-se eficaz na redução da obstrução e melhora clínica. O estudo reforça a importância de: Triagem familiar Diagnóstico precoce Seguimento rigoroso Abordagem multidisciplinar Conclusão A cardiomiopatia hipertrófica neonatal é uma condição rara, de difícil diagnóstico precoce e com potencial evolução grave. A ecocardiografia precoce associada à investigação genética e história familiar é essencial para diagnóstico e manejo. Mesmo com tratamento clínico, a doença pode progredir, sendo necessária intervenção invasiva em casos selecionados. O acompanhamento longitudinal e a triagem familiar são fundamentais para melhorar o prognóstico. Insights clínicos Quando suspeitar de cardiomiopatia hipertrófica em neonatos? Em casos de dispneia inexplicada, especialmente com história familiar positiva ou achados ecocardiográficos sugestivos. Qual exame é essencial para o diagnóstico? A ecocardiografia é o principal método, permitindo identificar hipertrofia miocárdica e obstrução do trato de saída. Qual a importância da história familiar? Fundamental, pois até 60% dos casos têm origem genética autossômica dominante. O tratamento clínico é suficiente? Nem sempre. Betabloqueadores podem retardar a progressão, mas casos graves podem necessitar intervenção invasiva. Quando considerar intervenção cirúrgica ou percutânea? Na presença de obstrução significativa do trato de saída e piora clínica progressiva. Qual o papel do teste genético? Confirma etiologia, orienta rastreio familiar e auxilia no prognóstico. Qual a principal complicação da CMH pediátrica? Morte súbita cardíaca, além de insuficiência cardíaca e arritmias. Para ver mais conteúdos como este, acesse: NeoPed Hub
Faça login para acessar o conteúdo
ou cadastre-se. | ESQUECI MINHA SENHA